Publications
The following is a list of my research publications.
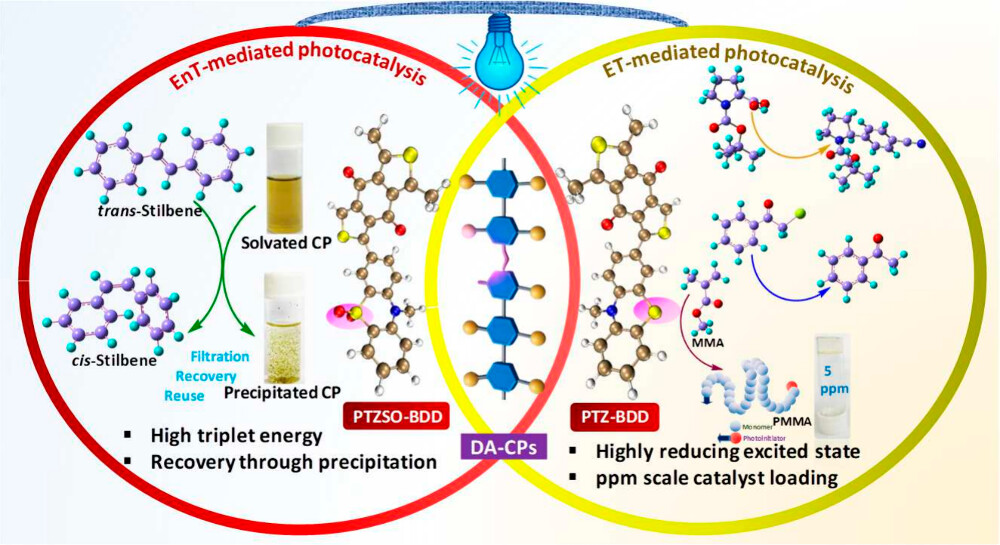
Abstract: Donor–acceptor (D–A) conjugated polymers (CPs) exhibit electron- and energy-transfer photocatalysis due to their accessible hybrid local and charge-transfer (HLCT) states, strong visible-light absorption, efficient charge separation, and notable photostability. Herein, we report that alkyl-chain-substituted phenothiazine- and benzodithiophenedione-based solution-processable CPs (PTZ-BDD and PTZSO-BDD), among which PTZSO-BDD shows efficient photocatalytic trans-to-cis isomerization of alkenes (up to 84% yield) within 2 h via triplet energy transfer from the photocatalyst to trans-alkenes. Effective recovery of these homogeneous catalysts via a reverse-dissolution process, achieved by heterogenization through solvent alteration, creates a platform that offers distinct advantages of both homogeneous and heterogeneous photocatalysts. Further, altering the chromogens in these CPs modulates the excitonic redox potential and frontier orbital alignment, enabling access to CT states that allow diverse photoredox catalysis such as hydrodebromination of phenacyl bromide, pinacol-type photochemical coupling of benzaldehyde, decarboxylative Giese-type conjugate addition, and dual metallophotoredox catalysis reactions, via oxidative and reductive quenching pathways. Femtosecond transient absorption spectroscopy revealed a prolonged absorption signal persisting up to 400 ps for PTZ-BDD, indicating a longer exitonic lifetime that enables efficient electron transfer from excited charge-transfer states. High chromophoric density in the polymeric backbone enables lower catalyst loading (up to 5 ppm) for the atom-transfer radical photopolymerization of methacrylates with high molecular weights. This work represents the first demonstration of accessing HLCT states by fine-tuning the donor and acceptor motifs of a solution-processable polymeric photocatalyst toward diverse and synthetically valuable photoredox organic transformations at exceptionally low catalyst loadings. Therefore, these metal-free photocatalysts not only demonstrated straightforward recycling but also meet crucial advantages of both homogeneous and heterogeneous catalysts, with low catalyst loading at the ppm level, improving economic sustainability.
TOC:
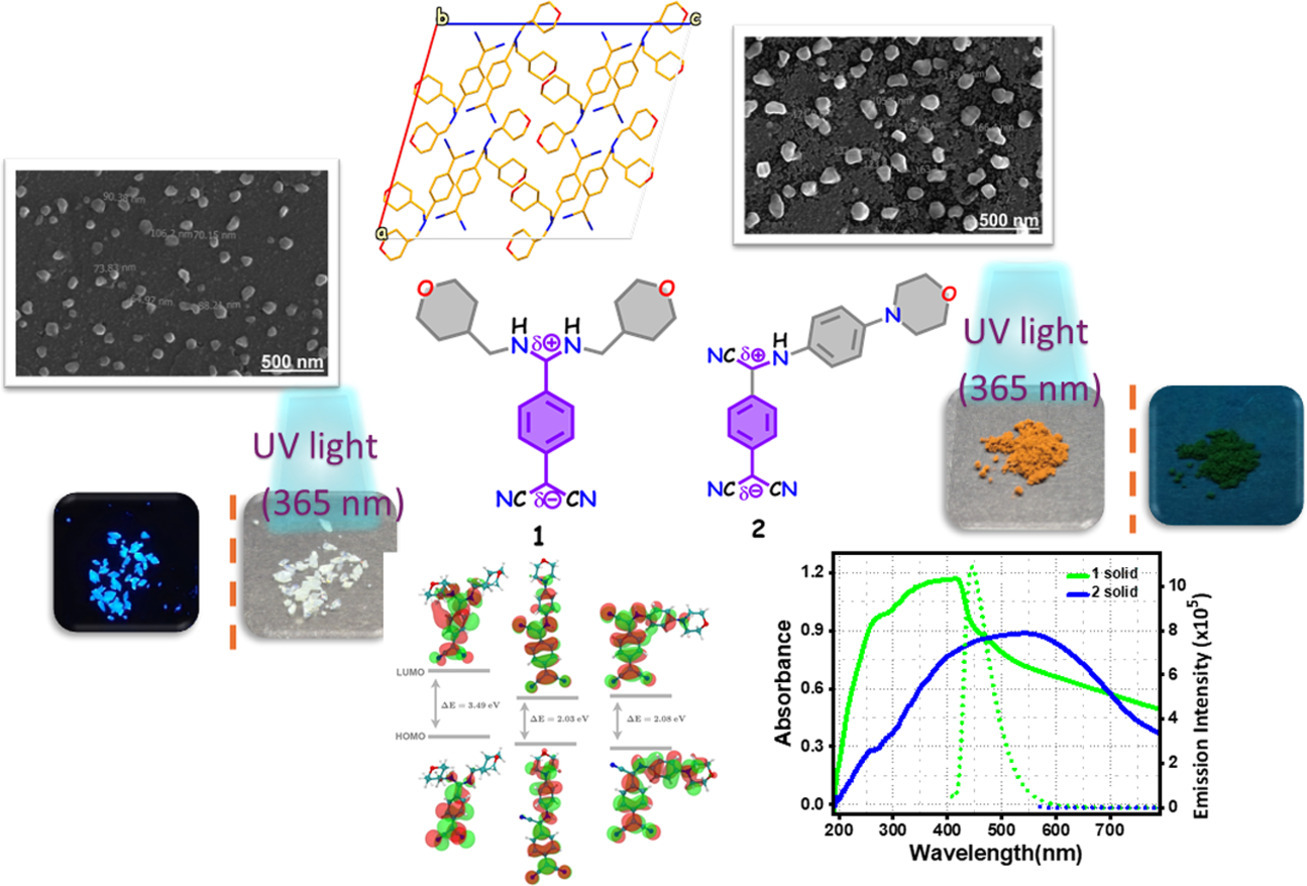
Abstract: Tetracyanoquinodimethane (TCNQ) derivatives, a disubstituted 7,7-bis(4-aminomethyltetrahydropyrano)-8,8-dicyanoquinodimethane (BAMTPDQ)1 and mono-substituted 7-(N-(4-aminophenyl)morpholino)-7-cyano-8,8-dicyanoquinodimethane (APMTCNQ) 2 were synthesised, illustrating structural, photophysical and thermal property variation with an aliphatic and an aromatic amine incorporated in the TCNQ derivatives, a design aspect. The absorption and emission wavelengths are red-shifted in both the solid and solution 2, owing to extended conjugation, compared to 1. Blue emissive solid 1 exhibits enhanced fluorescence; quantum yield (ϕf) ∼ 25.29 % due to supramolecular assembly with H-bonding and short contacts, restricting intermolecular motion, resulting in rigidity. On the contrary, solid 2 is non-emissive, possibly due to aggregation-caused quenching (ACQ). A typical semiconducting band gap was observed at 1 (∼2.4 eV) and 2 (∼1.7 eV). Electrochemically, 1, 2 undergo a diffusion-controlled redox mechanism and are thermally stable (∼280 °C). Drop-cast films of 1 and 2 exhibited distinct morphological features due to differences in growth kinetics. Computational analyses collectively suggest that the smaller HOMO–LUMO gap, greater orbital delocalization, and the absence of bulky nonplanar tetrahydropyran groups in 2 favour face-to-face π-π stacking, which may facilitate excitonic coupling and thereby increase the likelihood of ACQ, whereas 1 preferentially adopts antiparallel hydrogen-bonded assemblies that limit extensive stacking and help retain emissive behaviour. The morphology of drop-cast films showed nanoaggregates. Thus, the photophysical, electrochemical, and thermal properties, as well as the film-forming ability, of 1 and 2 demonstrate their suitability for organic electronic applications, such as organic photovoltaics (OPVs), organic light-emitting diodes (OLEDs), and liquid crystal displays (LCDs), among others.
TOC:
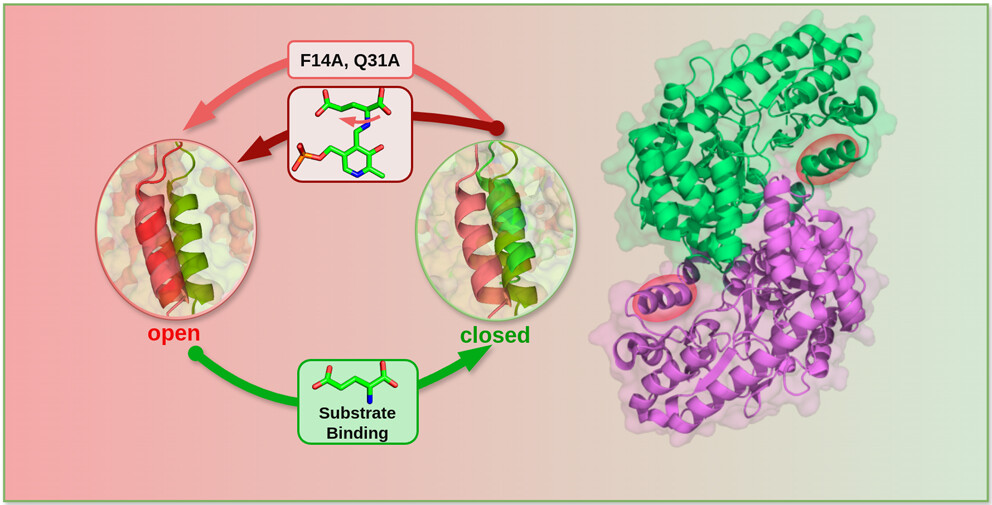
Abstract: The microbial aminotransferase enzyme DapC is vital for lysine biosynthesis in various Gram-positive bacteria, including Mycobacterium tuberculosis. Characterization of the enzyme’s conformational dynamics and identifying the key residues for ligand binding are crucial for the development of effective antimicrobials. This study employs atomistic simulations to explore and categorize the dynamics of DapC in comparison to other classes of aminotransferase. DapC undergoes an open-to-closed conformational change upon substrate binding, characterized by the movement of the N-terminal α2 helix, akin to that observed in the class Ib aspartate aminotransferase from Thermus thermophilus. Based on sequence similarity, essential dynamics, and the absence of the characteristic hinge movement, DapC is classified as a class Ib aminotransferase of type-I pyridoxal-5′-phosphate (PLP)-dependent enzyme. In the open state of DapC, two binding modes of glutamate, namely, canonical and alternate, separated by a dihedral rotation, are equally preferred. The closed state prefers the canonical binding mode, which is favorable for catalysis. In the case where the substrate binds in the alternate mode, a low-barrier dihedral rotation generates the canonical mode for efficient catalysis. The presence of two highly conserved residues, Phe14 and Gln31, stabilizes the closed state of substrate-bound DapC. Mutations of these residues disrupt the crucial hydrophobic interactions with the substrate, causing the enzyme to shift to an open state. While Phe14 has a dominant role, Gln31 is less consequential in regulating the conformational change, while the double mutation leads to a rapid conformation change.
TOC: